Thalassemia is an inherited blood disorder ( I.e. passed from parents to children through genes) where the body doesn’t make enough of a protein called hemoglobin.
The symptoms of Thalassemia vary depending on the type and severity of the condition:
Moderate to Severe Forms
Fatigue
Weakness
Pale or yellowish skin
Facial bone deformities
Slow growth
Loss of appetite
Dark urine
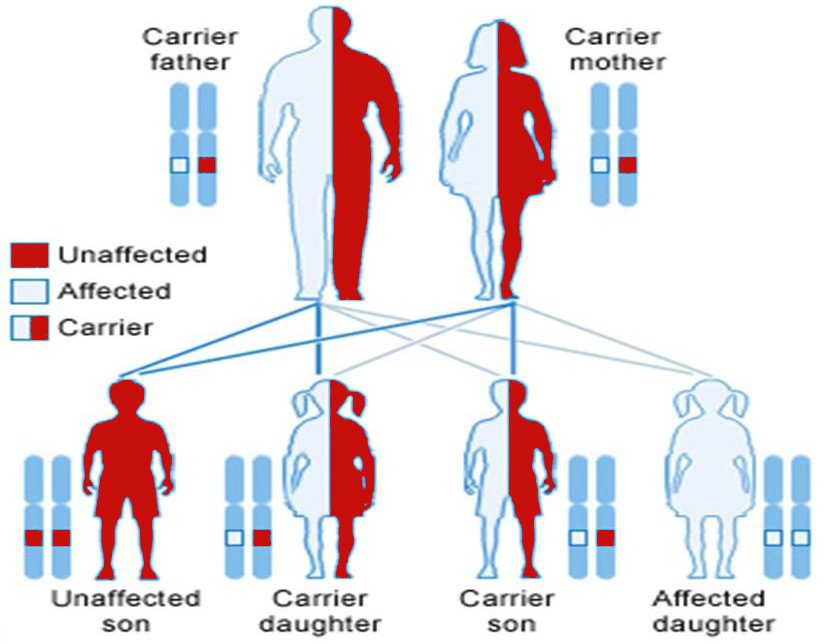
Some babies show signs and symptoms of Thalassemia at birth; others develop them during the first two years of life. Some people who have only one affected hemoglobin gene don’t have Thalassemia symptoms.
Moderate to severe Thalassemia can lead to various complications due to chronic anemia and the treatments required to manage the condition. Here are some possible complications:
Iron Overload
Frequent blood transfusions, a common treatment for Thalassemia, can lead to iron overload, where excess iron accumulates in the body.
Bone Deformities and Osteoporosis
Especially in the face and skull due to overactive bone marrow trying to produce more red blood cells.
Infections
People with Thalassemia, particularly those who have had their spleen removed, are at increased risk of infections due to a compromised immune system. Regular vaccinations and preventive antibiotics may be necessary.
Enlarged Spleen
The spleen may become enlarged as it works harder to filter out damaged red blood cells. Increased Anemia as the spleen destroys red blood cells more quickly. If the spleen becomes too large, there is a risk of rupture.
Moderate or severe Beta Thalassemia Major are often diagnosed in childhood because symptoms usually appear within the first two years of your child’s life. Your healthcare provider may order various blood tests to diagnose Thalassemia:
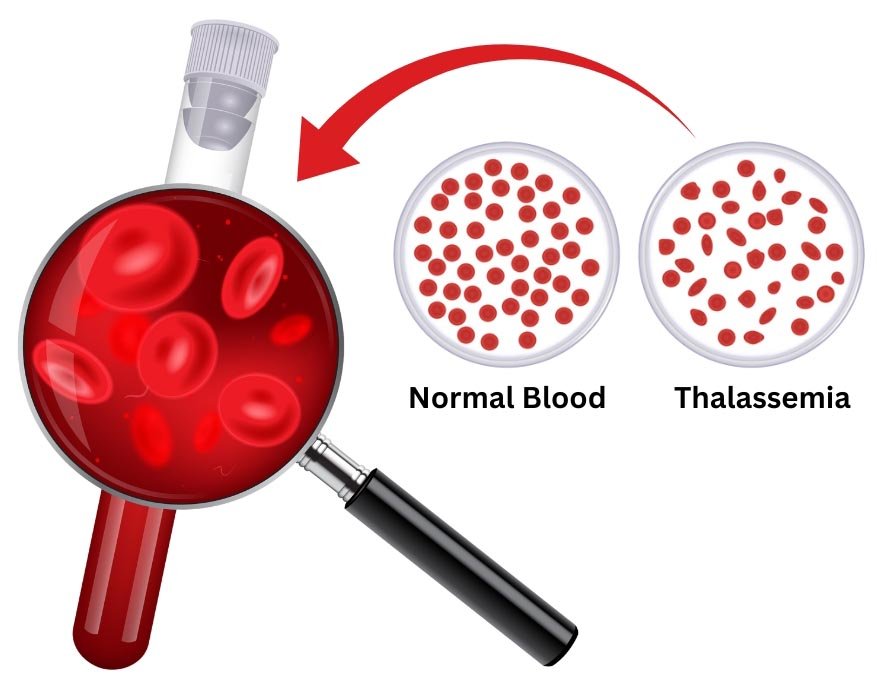
A complete blood count (CBC) that includes measures of hemoglobin and the quantity (and size) of red blood cells. People with Thalassemia have fewer healthy red blood cells and less hemoglobin than normal. They may also have smaller-than-normal red blood cells.
A reticulocyte count (a measure of young red blood cells) may indicate that your bone marrow isn’t producing enough red blood cells.
Studies of iron will indicate whether the cause of your anemia is an iron deficiency or Thalassemia.
Hemoglobin electrophoresis is used to diagnose beta Thalassemia
Genetic testing is used to diagnose alpha Thalassemia.
Standard treatments for Thalassemia major are blood transfusions and iron chelation.
A blood transfusion involves receiving injections of red blood cells through a vein to restore normal levels of healthy red blood cells and hemoglobin. You’ll receive transfusions every four months with moderate or severe Thalassemia, and with beta Thalassemia major, every two to four weeks. Occasional transfusions may be needed (for instance, during times of infection) for hemoglobin H disease or Beta Thalassemia intermedia.
Iron chelation involves the removal of excess iron from your body. A danger with blood transfusions is that they can cause iron overload. Too much iron may damage organs. If you receive frequent transfusions, you’ll receive iron chelation therapy (which you can take as a pill).
Folic acid supplements can help your body make healthy blood cells.
Bone marrow and stem cell transplant from a compatible related donor is the only treatment to cure Thalassemia. Compatibility means the donor has the same types of proteins, called human leukocyte antigens (HLA), on the surface of their cells as the person receiving the transplant. Your healthcare provider will inject bone marrow stem cells from your donor into your bloodstream during the procedure. The transplanted cells will start to make new, healthy blood cells within one month.